Алкилирование ароматических аминов. Прямое алкилирование аммиака и аминов
Существует огромное число разнообразных методов получения аминов. В этом разделе будут рассмотрены только наиболее общие и важные из них. Приведенные ниже способы синтеза аминов различаются областью своего применения, доступностью метода и количеством побочных продуктов при реализации требуемого превращения.
21.5.1.Прямое алкилирование аммиака и аминов
Амины получаются при взаимодействии первичных и вторичных алкилгалогенидов с аммиаком. Эта реакция была открыта А.Гофманом в 1849 году и является наиболее простым методом синтеза первичных, вторичных и третичных аминов, а также солей тетраалкиламмония. Реакция алкилгалогенидов с аммиаком или аминами относится к процессам бимолекулярного нуклеофильного замещения у насыщенного атома углерода, в которых аммиак или амин выполняют роль нуклеофильного агента. Переходное состояние такого процесса более полярно, чем исходные реагенты, поэтому скорость реакции резко возрастает в более полярной среде. В качестве растворителя обычно используют этанол или метанол, но более эффективны диполярные апротонные растворители ДМФА, ДМАА. Алкилирование аммиака с целью получения аминов нашло широкое применение в промышленности, но все реже и реже используется в лабораторных условиях, поскольку в этой реакции всегда образуется смесь первичного, вторичного и третичного амина, а при наличии избытка алкилгалогенида и соли тетраалкиламмония.
Катион алкиламмония, как было отмечено выше, обладает свойствами слабой кислоты. В результате переноса протона к молекуле аммиака образуется первичный амин и катион аммония. Первичный амин проявляет свойства более сильного нуклеофильного агента, чем аммиак, и при взаимодействии с алкилгалогенидами дает катион диалкиламмония, из которого далее получается вторичный амин. Этот процесс может продолжаться далее, приводя к третичному амину и даже к соли тетраалкиламмония. Вся последовательность происходящих превращений описывается приведенными выше уравнениями (1)-(7). Соотношение продуктов реакции зависит от соотношения исходных реагентов. Увеличение количества алкилгалогeнида способствует росту доли третичного амина и четвертичной аммониевой соли, в то время как в присутствии избытка аммиака преимущественно образуется смесь первичного и вторичного амина. Однако даже при большом избытке аммиака реакцию невозможно остановить на стадии образования только первичного амина. В типичном примере взаимодействия одного моля 1-бромоктана и трех молей аммиака при 20°С получается смесь, состоящая из 45% октиламина, 43% диоктиламина и следов триоктиламина. При большем количестве аммиака доля первичного амина возрастает, но вторичный амин всегда присутствует в продуктах реакции.

Таким образом, прямое алкилирование оказывается малоудовлетворительным методом для получения чистых первичных, вторичных и третичных аминов.
21.5.2.Непрямое алкилирование. Синтез первичных аминов по Габриэлю
В 1887 году Габриэль предложил простой и очень удобный общий метод получения первичных аминов. Фталимид калия алкилируется под действием алкилгалогенидов с образованием N-алкилфталимида с очень высоким выходом.


Гидразин является наилучшим реагентом для снятия фталоильной защиты с атома азота. Ранее для этой цели использовали щелочной или кислотный гидролиз. Фталимид получается в промышленных условиях при взаимодействии фталевой кислоты или ее ангидрида с газообразным аммиаком при 300°-350°С. Фталимид представляет собой средней силы N-H кислоту с рК а ~ 8,3. При взаимодействии фталимида с гидроксидом калия в водно-спиртовой среде получается К-соль фталимида. Синтез Габриэля можно рассматривать как один из лучших способов получения первичных аминов из первичных и вторичных но не третичных алкилгалогенидов. Этот метод широко используется также и для получения эфировa-аминокислот.



В качестве примера применения реакции Габриэля для получения первичных аминов приведем синтез дофамина - важного синтетического регулятора деятельности центральной нервной системы.

КУРСОВАЯ РАБОТА
по дисциплине: «Дополнительные главы органической химии»
на тему: «Ацилирование и алкилирование аминов»
1. Введение ………………………………………………………………… 3
2. Ацилирование и алкилирование аминов ………………………………. 5
2.1. Амины: номенклатура, классификация, применение …………… 5
2.2. Химические свойства аминов ……………………………………... 9
2.2.1. Основные и кислотные свойства ……………………………. 9
2.2.2. Реакции ацилирования ……………………………………… 12
2.2.3. Реакции алкилирования …………………………………….. 17
2.2.4. Ацилирование и алкилирование по Фриделю-Крафтсу ….. 21
2.2.5. Взаимодействие аминов с азотистой кислотой …………… 25
2.3. Методы получения аминов ………………………………………. 28
2.3.1. Алкилирование аммиака и аминов ………………………… 28
2.3.2. Восстановление азотсодержащих органических соединений …………………………………………………... 29
2.3.3. Перегруппировка Гофмана …………………………………. 30
2.4. Биологически активные амины и их производные …………….. 30
2.5. Акридон: получение, свойства и применение ………………….. 36
2.6. 9-аминоакридин: получение, свойства и применение …………. 38
3. Заключение …………………………………………………………….. 39
4. Список литературы …………………………………………………….. 40
1. Введение
Амины производные аммиака, в молекуле которого один, два или три атома водорода заметены углеводородными радикалами. По числу замешенных атомов водорода они делятся на первичные, вторичные и третичные амины.
Существуют и четвертичные аммониевые соли, например хлорид тетраметиламония, соответствующее ему основание гидроксид тетраметиламмония, который представляет собой сильное основание, аналогичное щелочных металлов, так как связь с гидроксильной группой здесь ионная .
В зависимости от природы углеводородных радикалов амины подразделяются на алифатические, алициклические, ароматические и смешанные (имеющие алифатический и ароматический радикалы). Эти соединения отличаются друг от друга строением углеводородных радикалов.
Названия простых по строению аминов образуют от названий соответствующих углеводородных радикалов, связанных с атомом азота, добавляя в конце корень-амин. Кроме того, ароматические амины имеют тривиальные названия.
Нуклеофильность и основность аминов изменяются, как правило, симбатно: они уменьшаются с уменьшением электроннной плотности на атоме азота или при его пространственном экранировании и увеличиваются с увеличением электронной плотности на атоме азота или с увеличением его доступности.
Широко амины и их производные применяется в производстве инсектицидов, фунгицидов, ускорителей вулканизации, поверхностно-активных веществ, красителей, ракетных топлив, растворителей и т.д.
Все выше сказанное подтверждает актуальность темы представленной работы.
Цель работы: изучить реакции ацилирования и алкилирования аминов.
Для достижения поставленной цели были определены следующие задачи:
1. Рассмотреть общую характеристику аминов.
2. Рассмотреть химические свойства аминов.
3. Проанализировать методы получения аминов.
4. Дать характеристику биогенным аминам.
5. Представить получение, свойства и применение 9-аминоакридина.
2. Ацилирование и алкилирование аминов
2.1. Амины: номенклатура, классификация, применение
Аминами называются органические производные аммиака, в котором один, два или все три атома водорода замещены на углеводородные радикалы (предельные, непредельные, ароматические).
Название аминов производят от названия углеводородного радикала с добавлением окончания -амин или от названия соответствующего углеводорода с приставкой амино-.
Примеры приведена на Рис. 1:
CH 3 NH 2 CH 3 NH C 2 H 5
Метиламин метилэтиламин метилдифениламин
Фениламин (анилин)
Рис. 1. Примеры полуструктурных формул аминов и их названия
В зависимости от числа атомов водорода, замещенных в аммиаке на углеводородные радикалы, различают первичные, вторичные и третичные амины (Рис. 2):
R - NH 2 R NH R R N R ”
первичный амин вторичный амин третичный амин
Рис. 2. Классификация аминов. R , R , R углеводородные радикалы
Алкиламины содержат только алифатические углеводородные радикалы, например (Рис. 3):
Рис. 3. Примеры алкиламинов
Ариламины содержат ароматические радикалы с атомом азота в ароматическом кольце, например (ри№с. 4):
Рис. 4. Примеры ариламинов
Алкилариламины содержат алифатические и ароматические радикалы, например (Рис. 5):
Рис. 5. Примеры алкилариламинов
Гетероциклические амины содержат азот в цикле, например (Рис. 6):
Рис. 6. Примеры гетероциклических аминов
Амины, чаще в виде полифункциональных производных, находят применение, являясь обычно полупродуктами в органических синтезах. Получаются с применением аминов такие лекарственные препараты, как новокаин, спазмолитин, парацетамол, сульфаниламидные препараты .
Широкое применение нашли соединения с упрощенной адреналиновой структурой, такие, как эфедрин, амфетамин, первитин и т.д. (Рис. 7)
Рис. 7. Формулы эфедрина (а), амфетамина (b ), первитина (с)
Эти соединения, обладая структурой близкой к структуре адреналина, оказывают стимулирующее, возбуждающее действие, но более сильное и продолжительное.
Амины широко используются в качестве термо- и светостабилизаторов (Рис. 8):
Рис. 8. Формулы представителей аминов термо- и светостабилизаторов
Амины могут выступать в роли модификаторов резин и как вулканизирующие агенты (Рис. 9):
Рис. 9. Формулы представителей аминов модификаторов резин и вулканизирующие агенты
Мономеры для синтеза полиамидов (Рис. 10):
Рис. 10. Формулы представителей аминов
мономеров для синтеза полиамидов
Амины могут быть использованы и как красители (Рис. 11):
Рис. 11. Формулы представителей аминов красителей
Фотореактивы (Рис. 12):
Рис. 12. Формулы представителей аминов фотореактивов
Метиламин применяется в производстве инсектицидов, фунгицидов, ускорителей вулканизации, поверхностно-активных веществ, красителей, ракетных топлив, растворителей.
Триэтиламин применяется в производстве ускорителей вулканизации, ингибиторов коррозии, растворитель.
Анилин: производство N,N-диметиланилина, дифениламина, лекарственных средств, антиоксидантов, ускорителей вулканизации и фотоматериалов.
Некоторые амины применяются как селективные растворители для извлечения урана из сернокислых растворов. Амины, обладающие запахом рыбы, используются как приманка в борьбе с полевыми грызунами.
Третичные амины и соли четвертичных аммониевых оснований получили широкое распространение в качестве катализаторов межфазного переноса в органическом синтезе .
2.2. Химические свойства аминов
Химические свойства аминов определяются в основном присутствием атома азота с неподеленной парой электронов, наличие которой обуславливает их основные и нуклеофильные свойства.
2.2.1. Основные и кислотные свойства
Алифатические амины являются сильными основаниями (= 10-11) и превосходят по основности аммиак. Их водные растворы имеют щелочную реакцию:
RNH 2 + H 2 O → RNH 3 + + OH (1)
Ароматические амины слабые основания (= 3-5), что связано с разрушением при протонированиии стабильной сопряженной системы, в которой участвует неподеленная пара электронов азота (см. лек. №4).
При взаимодействии с кислотами амины образуют растворимые в воде аммониевые соли:
RNH 2 + HX → RNH 3 + X - (2)
Первичные и вторичные амины являются слабыми N-H кислотами (рК а =33-35) и образуют соли при взаимодействии с активными металлами:
RNH 2 + Na → RNH - Na + + 1/2 H 2 (3)
Характер групп, находящихся у атома азота, оказывает большое влияние на основность амина. Обычно алифатические амины, являются сильными основаниями, обладают щелочной реакцией на лакмус и во влажном состоянии поглощают двуокись углерода. Низшие алифатические амины являются, более сильными основаниями, чем аммиак, и титруются кислотами в присутствии метилоранжа или бромфенолблау в качестве индикатора. При наличии ароматического остатка основность аминов выражена значительно слабее; например, анилин и его гомологи, хотя и образуют соли с разбавленными минеральными кислотами, однако не дают щелочной реакции на лакмус и не поглощают двуокись углерода из воздуха. Титрование таких аминов кислотой в присутствии обычных индикаторов не дает удовлетворительных результатов. Напротив, солянокислые соли ароматических аминов легко титруются водным раствором щелочи в присутствии фенолфталеина, т.е. ведут себя в этих условиях, как свободные кислоты. При увеличении числа ароматических радикалов у атома азота наблюдается еще большее уменьшение основности амина. Соли дифениламина гидролизуются в воде в значительной степени с частичным выделением свободного основания. Трифениламин является нейтральным соединением и не образует солей, за исключением комплексного соединения с хлорной кислотой.
Хотя введение нитрогруппы в ядро анилина заметно понижает его основность, нитранилины все же еще обладают основным характером и дают соли с минеральными кислотами, которые, однако, очень легко, гидролизуются при действии воды. Введение более одной нитрогруппы в ядро ведет к дальнейшему понижению основности; действительно, иолинитрамины проявляют лишь незначительную склонность к образованию солей. То же относится и к галоидозамещенным аминам .
Обычно, для получения соли к амину прибавляют небольшой избыток кислоты, например разбавленной серной, концентрированной или разбавленной соляной или бромистоводородной кислоты. Если при этом соль не выпадает, раствор упаривают на водяной бане или в вакуум-эксикаторе до начала кристаллизации. Если галоидоводородные соли плохо кристаллизуются из водного раствора, их получают другим путем, а именно пропусканием сухого галоидоводорода в раствор амина в бензоле, хлороформе или эфире. Этот метод особенно удобен для получения галоидоводородных солей алкиланилинов и диалкиланилинов, а также аминов, соли которых легко гидролизуются водой. При пропускании сухого хлористого водорода в раствор диалкиланилина в сухом эфире до насыщения соответствующие солянокислые соли легко выделяются в кристаллическом состоянии. При этом следует тщательно предохранять реакционную смесь от доступа влаги. Солянокислые соли низших алкиланилинов лучше всего получать в бензольном растворе. Впрочем, в случае алкиланилинов, содержащих сравнительно большие алкильные радикалы, этот способ не дает таких удовлетворительных результатов.
Большинство аминов образует хорошо кристаллизующиеся пикраты, которые могут служить для идентификации аминов или для выделения их из смесей. Обычно, пикраты получаются смешением обоих компонентов в подходящем растворителе, выбор которого определяется сравнительной растворимостью в нем пикриновой кислоты, пикрата и амина. Менее удобно пользоваться для этой цели реакцией обмена. Пикролоновая кислота также применяется для идентификации аминов, особенно в тех случаях, когда пикриновая кислота не дает удовлетворительных результатов. Соли пикролоновой кислоты обычно труднее растворимы, чем пикраты, и обладают более высокой температурой плавления. Этот способ применяется главньм образом для идентификации простейших алифатических производных гидроксиламина, производных морфолина и некоторых алкалоидов. Кроме того, для идентификации аминов также применяется имидазолдикарбоновая кислота.
Ароматические амины образуют продукты присоединения с ди- и тринитросоединениями. Эти продукты также иногда служат для идентификации аминов. Некоторые амины при действии 70% водной хлорной кислоты дают хорошо кристаллизующиеся соли, которые могут служить для их выделения и идентификации .
2.2.2. Реакции ацилирования
Ацилирование введение ацильной группы (ацила) RC в молекулу органического соединения путем замещения атома водорода. В широком смысле ацилирование это замещение любого атома или группы атомов на ацила. В зависимости от атома к которому присоединяют ацил различают C-, N-, O-, S ацилирование.
Реакции ацилирования обладают очень многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). Таким образом, можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и другие полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях. В своей курсовой работе я рассмотрю три наиболее важных типа реакций ацилирования C-ацилирование, O-ацилирование и N-ацилирование.
Ацилирование аминов ацилгалогенидами
Реакции аминодегалогенирования наиболее часто используются для синтеза амидов. Действие аммиака или аминов на ацилгалогениды представляет собой общий метод синтеза амидов (Рис. 13):
Рис. 13. Общий метод синтеза амидов
Реакция сильно экзотермична и требует тщательного контроля, обычно охлаждением или разбавлением. При использовании аммиака получают незамещенные амиды, из первичных аминов получают N-замещенные амиды, а из вторичных аминов N, N-дизамещенные амиды. Аналогично можно ацилировать ариламины. В некоторых случаях для связывания выделяющейся галогеноводородной кислоты добавляют водный раствор щелочи. Такая реакция носит название метода Шоттена-Баумана.
Гидразин и гидроксиламин также реагируют с ацилгалогенидами, давая соответственно гидразиды RCONHNH 2 и гидроксамовые кислоты RCONHOH; эта реакция часто используется для синтеза данных соединений. Если вместо ацилгалогенида взять фосген, то как ароматические, так и алифатические первичные амины дают хлороформамиды ClCONHR, которые теряя HCl, превращаются в изоцианаты RNCO. Это один из наиболее распространенных методов синтеза изоцианатов.
Тиофосген при аналогичной обработке дает изотиоцианаты. Фосген в этой реакции можно заменить более безопасным трихлорометилхлороформиатом. При действии первичных аминов на хлороформиаты ROCOCl получаются карбаматы ROCONHR. Примером этой реакции служит защита аминогруппы в аминокислотах и пептидах действием карбобензоксихлорида (Рис. 14):
Рис. 14. Защита аминогруппы в аминокислотах и пептидах
действием карбобензоксихлорида
Аминогруппы вообще часто защищают превращением в более устойчивые амидные. Взаимодействие ацилгалогенидов с нитридом лития дает N, N-диациламиды (триациламины).
Ацилирование аминов ангидридами
По механизму и диапазону применимости реакция амино-деацилоксизамещения может быть проведена с участием аммиака, первичных или вторичных аминов (Рис. 15)
Рис. 15. Реакция аминодеацилоксизамещения
Однако при использовании аммиака и первичных аминов получаются также и имиды, в которых с атомом азота связаны две ацильные группы. Это происходит особенно легко в случае циклических ангидридов, из которых образуются циклические амиды (Рис. 16):
Рис. 16. Реакция
получения имидов
Второй стадией этой реакции, которая намного медленнее первой, является атака атома азота амидной группы на карбоновую кислоту.
Ацилирование аминов карбоновыми кислотами
При обработке карбоновых кислот аммиаком или аминами получаются соли. Соли, полученные из аммиака, а также первичных и вторичных аминов в результате пиролиза дают амиды, но этот метод менее удобен, чем реакции аминов с ангидридами, ацилгалогенидами и сложными эфирами, и редко используется в препаративных целях .
Хотя и взаимодействие кислот с аминами не приводит непосредственно к амидам, можно добиться чтобы эта реакция шла с хорошим выходом при комнатной или немного повышенной температуре (Рис. 17):
Рис. 17. Реакция получения амидов
Кислоты можно превратить в амиды также нагреванием с амидами других карбоновых кислот (обмен), сульфоновых или фосфиновых кислот или действием трис(алкиламино) боранов или трис(диалкиламино) боранов (Рис. 18):
Рис. 18. Превращение кислот в амиды
Ацилирование аминов сложными эфирами
Превращение сложных эфиров в амиды полезный метод синтеза незамещенных, N-замещенных и N, N-дизамещенных амидов из соответствующих аминов (Рис. 19):
Рис. 19. Превращение сложных эфиров в амиды
Реакцию можно проводить с алкильными или ароматическими группами R и R. Особенно хорошей уходящей группой является n-нитрофенильная. Эта реакция весьма ценна, так как многие сложные эфиры легкодоступны или сравнительно легко получаются даже в тех случаях, когда этого нельзя сказать о соответствующем ангидриде кислоты или ацилгалогениде. Как и по реакции с ацилгалогенидами, этим методом из сложных эфиров можно синтезировать гидразиды и гидроксамовые кислоты действием гидразина и гидроксиламина соответственно. И гидразин, и гидроксиламин взаимодействуют быстрее, чем аммиак или первичные амины. Вместо сложных эфиров часто используют фенилгидразиды, получаемые из фенилгидразина.
Остаётся добавить, что реакция образования гидроксамовых кислот. которые в присутствии трёхвалентного железа дают окрашенные комплексы, часто используется как тест на сложные эфиры.
Ацилирование аминов амидами
Это реакция обмена, и ее обычно проводят с солью амина. Уходящей группой служит, как правило, NH 2 , а не NHR или NR 2 ; в качестве реагентов наиболее широко применяются первичные амины (в виде солей).
Для образования комплекса с уходящим аммиаком можно добавлять BF 3 . Эту реакцию часто применяют для получения замещенных производных мочевины из самой мочевины (Рис. 20):
Рис. 20. Ацилирование аминов амидами
Диметилформамид можно превратить в другие формамиды продолжительным нагреванием с первичным или вторичным амином (Рис. 21) :
Рис. 21. Превращение диметилформамида в другие формамиды
2.2.3. Реакции алкилирования
N -алкилирование нередко классифицируют как аммонолиз (или аминолиз) органических соединений).
Алкилирование по атомам других элементов (Si -, Pb -, AI -алкилирование) представляет собой важнейший путь получения эл е мент- и металлорганических соединений, когда алкильная группа неп о средственно связывается с гетероатомом (Рис. 22):
2 RCI + Si R 2 SiCI 2
4 C 2 H 5 CI + 4 PbNa → Pb (C 2 H 5 ) 4 + 4 NaCI + 3 Pb
3 C 3 H 6 + AI + 1,5 H 2 → Al (C 3 H 7 ) 3
Рис. 22. Реакции алкилирования
Другая классификация реакций алкилирования основана на разл и чиях в строении алкильной группы, вводимой в органическое или нео р ганическое соединение. Она может быть насыщенной алифатической (этильной и изопропильной) или циклической. В последнем случае р е акцию иногда называют циклоалкилированием (Рис. 23):

Рис. 23. Реакция циклоалкилированием
При введении фенильной или вообще арильной группы образуется непосредственная связь с углеродным атомом ароматического ядра (арилирование) (Рис. 24):
C 6 H 5 CI + NH 3 → C 6 H 5 NH 2 + HCI
Рис. 24. Арилирование
В алкильную группу может входить ароматическое ядро или дво й ная связь, и, если последняя достаточно удалена от реакционного це н тра, реакция мало отличается от обычных процессов алкилирования (Рис. 25):
CH 2 =CH-CH 2 CI + RNH 2 → RNHCH 2 -CH=CH 2 + HCI
Рис. 25. Реакция алкилирования
Однако введение винильной группы (винилирование) занимает ос о бое место и осуществляется главным образом при помощи ацетилена (Рис. 26):
ROH + CH≡CH ROCH=CH 2
CH 3 -COOH + CH≡CH CH 3 -COO-CH=CH 2
Рис. 26. Реакция винилирования
Наконец, алкильные группы могут содержать различные заместит е ли, например атомы хлора, гидрокси-, карбокси-, сульфокислотные группы (Рис. 27):
C 6 H 5 ONa + CICH 2 -COONa → C 6 H 5 O-CH 2 -COONa + NaCI
ROH + HOCH 2 -CH 2 SO 2 ONa → ROCH 2 CH 2 SO 2 ONa + H 2 O
Рис. 27. Структура алкильных групп
Важнейшей из реакций введения замещенных алкильных групп я в ляется процесс β-оксиалкилирования (в частном случае оксиэтилир о вание), охватывающий широкий круг реакций оксидов олефинов (Рис. 28):
Рис. 28. Оксиэтилир о вание
Алкилирующие агенты и катализаторы
Все алкилирующие агенты по типу связи, разрывающейся в них при алкилировании, целесообразно разделить на следующие группы:
1. Ненасыщенные соединения (олефин и ацетилен), у которых пр о исходит разрыв -электронной связи между атомами углерода;
2. Хлорпроизводные с достаточно подвижным атомом хлора, сп о собным замещаться под влиянием различных агентов;
3. Спирты, простые и сложные эфиры, в частности оксиды олеф и нов, у которых при алкилировании разрывается углерод-кислородная связь.
Олефины (этилен, пропилен, бутены и высшие) имеют первост е пенное значение в качестве алкилирующих агентов. Ввиду дешевизны ими стараются пользоваться во всех случаях, где это возможно. Гла в ное применение они нашли для С-алкилирования парафинов и аром а тических соединений. Они неприменимы для N -алкилирования и не вс е гда эффективны при S - и O -алкилировании и синтезе металлорганич е ских соединений .
Алкилирование олефинами в большинстве случаев протекает по ионному механизму через промежуточное образование карбокатионов и катализируется протонными и апротонными кислотами. Реакционная способность олефинов при реакциях такого типа определяется их склонностью к образованию карбокатионов:
(4)
Это означает, что удлинение и разветвление цепи углеродных ат о мов в олефине значительно повышает его способность к алкилиров а нию:
CH 2 =CH 2 < CH 3 -CH=CH 2 < CH 3 -CH 2 -CH=CH 2 < (CH 3 ) 2 C=CH 2 (5)
В ряде случаев алкилирование олефинами протекает под влиянием инициаторов радикально-цепных реакций, освещения или высокой те м пературы. Здесь промежуточными активными частицами являются св о бодные радикалы. Реакционная способность разных олефинов при т а ких реакциях значительно сближается.
Хлорпроизводные являются алкилирующими агентами наиболее широкого диапазона действия. Они пригодны для С-, О-, S - и N -алкилирования и для синтеза большинства элементо- и металлорган и ческих соединений. Применение хлорпроизводных рационально для тех процессов, в которых их невозможно заменить олефинами или когда хлорпроизводные дешевле и доступнее олефинов.
Алкилирующее действие хлорпроизводных проявляется в трех ра з личных типах взаимодействий: в электрофильных реакциях, при ну к леофильном замещении и в свободно-радикальных процессах. Мех а низм электрофильного замещения характерен для алкилирования по атому углерода, но, в отличие от олефинов, реакции катализируются только апротонными кислотами (хлориды алюминия, железа). В пр е дельном случае процесс идет с промежуточным образованием карбок а тиона:
(6)
в связи, с чем реакционная способность алкилхлоридов зависит от п о ляризации связи C - CI или от стабильности карбокатионов и повышается при удлинении и разветвлении алкильной группы:
(7)
При другом типе реакций, характерном для алкилирования по ат о мам кислорода, серы и азота, процесс состоит в нуклеофильном зам е щении атома хлора. Механизм аналогичен гидролизу хлорпроизводных, причем реакция протекает в отсутствие катализаторов:
(8)
Реакционная способность хлорпроизводных изменяется в данных процессах таким же образом, как при гидролизе, а именно:
ArCH 2 CI > CH 2 =CH-CH 2 CI > AIkCI > ArCI (9)
перв -AIkCI > втор -AIkCI > трет -AIkCI (10)
Целый ряд процессов алкилирования хлорпроизводными протекает по свободно-радикальному механизму. Это особенно характерно для синтезов элементо- и металлорганических соединений, когда свободные радикалы образуются за счет взаимодействия с металлами :
4 PbNa + 4 C 2 H 5 CI → 4 Pb + 4 NaCI + 4 C 2 H → 4 NaCI + Pb (C 2 H 5 ) 4 + 3 Pb (11)
2.2.4.
Ацилирование и алкилирование по Фриделю-Крафтсу
Ацилирование по Фриделю-Крафтсу
Введение ацильной группы в ароматическое кольцо с помощью ацилирующего агента и кислоты Льюиса называют ацилированием по Фриделю-Крафтсу. Ацилирующими агентами обычно являются галогенангидриды и ангидриды кислот в присутствии галогенидов алюминия, трифторида бора или пентафторида сурьмы в качестве кислот Льюиса. Ацилгалогениды и ангидриды кислот образуют с кислотой Льюиса донорно-акцепторные комплексы состава 1:1 и 1:2. Спектральными методами было установлено, что хлорид алюминия, трифторид бора и пентафторид сурьмы координируются по карбонильному атому кислорода, так как он более основен чем соседний атом хлора. Электрофильным агентом в реакции ацилирования ароматических соединений является либо этот донорно-акцепторный комплекс, либо катион ацилия, образующийся при его диссоциации (Рис. 29).
Рис. 29. Реакция ацилирования по Фриделю-Крафтсу
Можно полагать, что медленной стадией реакции является атака одного из трех электрофилов на арен, приводящая к σ -комплексу. Эффективность этих ацилирующих частиц зависит от природы субстрата, ацилгалогенида и растворителя, а также от количества взятого катализатора.
При ацилировании аренов ацилгалогенидами, катализируемом хлоридом или бромидом алюминия в полярных апротонных растворителях (нитробензоле, нитрометане и др.), ацилирующим агентом является катион ацилия, тогда как в малополярной среде (хлористом метилене, дихлорэтане или тетрахлорэтане) в реакции принимает участие донорно-акцепторный комплекс. Природа ацилгалогенида также оказывает влияние на образование и стабильность солей ацилия. Механизм реакции ацилирования аренов по Фриделю-Крафтсу под действием донорно-акцепторного комплекса (Рис. 30):

Рис. 30. Механизм реакции ацилирования аренов по Фриделю-Крафтсу
Ароматический кетон представляет собой более сильное основание Льюиса, чем ацилгалогенид и образует стабильный комплекс с AlCl 3 или другой кислотой Льюиса. Поэтому для ацилирования ароматических соединений ацилгалогенидами требуется несколько больше эквимолярного количества катализатора, а при ацилировании ангидридами кислот два моля катализатора (т.к. они содержат два карбонильных атома кислорода). Кетон выделяют, разлагая его комплекс с AlCl3 водой или соляной кислотой.
Ацилирование по Фриделю-Крафтсу полностью лишено тех недостатков, которые присущи реакции алкилирования. При ацилировании вводится только одна ацильная группа, поскольку ароматические кетоны не вступают в дальнейшую реакцию (так же, как и другие арены, содержащие сильные электроноакцепторные группы: NO 2 , CN, COOR). Еще одним преимуществом этой реакции по сравнению с алкилированием является отсутствие перегруппировок в ацилирующем агенте. Кроме того, для ацилирования не характерны реакции диспропорционирования продуктов реакции .
Алкилирование по Фриделю-Крафтсу
Реакция Ш.Фриделя-Дж.Крафтса (1877 г.) представляет собой удобный метод прямого введения алкильной группы в ароматическое кольцо. Алкилирование ароматических соединений осуществляется под действием алкилгалогенидов, только в присутствии в качестве катализатора подходящей кислоты Льюиса: AlBr 3 , AlCl 3 , GaBr 3 , GaCl 3 , BF 3 , SbF 5 , SbCl 5 , FeCl 3 , SnCl 4 , ZnCl 2 и др. (Рис. 31):
Рис. 31. Алкилирование ароматических соединений
Наиболее активными катализаторами являются безводные сублимированные бромиды алюминия и галлия, пятифтористая сурьма, хлориды алюминия и галлия, менее активны галогениды железа (III), SbCl5, к малоактивным катализаторам относятся SnCl 4 и ZnCl 2 . В целом активность кислот Льюиса, как катализаторов алкилирования бензола, уменьшается в ряду AlBr 3 >GaBr 3 >AlCl 3 >GaCl 3 >FeCl 3 >SbCl 5 >TiCl 4 >BF 3 >BCl 3 >SnCl 4 >SbCl 3 . Самым распространенным катализатором этой реакции является предварительно сублимированный хлористый алюминий.
Например, механизм реакции бензилирования хлористым бензилом в нитробензоле в присутствии безводного AlCl 3 в качестве катализатора следующей схемой (Рис. 32):
Рис. 32. Механизм реакции бензилирования, где В: =AlCl 4 -; H 2 O или др. основание. Скорость реакции лимитируется второй стадией
Точное строение интермедиата неизвестно. В принципе, можно представить целый ряд структур от молекулярного комплекса до диссоциированных карбокатионов (Рис. 33):
Рис. 33. Строение интермедиата
Участие свободных карбокатионов как алкилирующих агентов маловероятно.
Если бы алкилирующими агентами были свободные карбокатионы, то медленной стадией была бы стадия их образования (k 1 ), а реакция с аренами была бы быстрой и третьего порядка не должно было наблюдаться. Крайне маловероятно, что алкилирующим агентом является молекулярный комплекс. При низких температурах иногда удается выделить комплексы алкилгалогенидов с кислотами Льюиса. Для них характерен медленный обмен галогенов по схеме (Рис. 34):
Рис. 34. Получение комплексов алкилгалогенидов с кислотами Льюиса
Скорость обмена возрастает в ряду перв.R< втор.R<трет.R, что можно объяснить и ион-парным строением, и структурой координационного аддукта.
Многие исследователи, работающие в данной области, полагают, что строение комплексов RX. MXn постепенно изменяется от структуры координационного аддукта в случае R=СН 3 до структуры ионной пары в случае R=t-Bu, однако экспериментально это пока не подтверждено.
Способность атома галогена в RX к комплексообразованию с AlCl 3 или другой жесткой кислотой Льюиса резко уменьшается от фтора к иоду, вследствие этого активность алкилгалогенидов в качестве алкилирующих агентов в реакции Фриделя-Крафтса также уменьшается в ряду RF>RCl>RBr>RI. По этой причине алкилиодиды не применяют в качестве алкилирующего агента.
Различие в активности алкилфторидов и алкилбромидов настолько велико, что позволяет селективно замещать фтор в присутствии брома в одной и той же молекуле (Рис. 35) :
Рис. 35. Различие в активности алкилфторидов и алкилбромидов
2.2.5. Взаимодействие аминов с азотистой кислотой
Первичные, вторичные и третичные амины по-разному взаимодействуют с азотистой кислотой, что используется для установления типа амина. Неустойчивую азотистую кислоту генерируют действием сильной кислоты на нитриты.
Третичные алифатические амины при обычной температуре с азотистой кислотой не взаимодействуют.
Вторичные амины образуют с азотистой кислотой устойчивые нитрозамины жидкие или твердые продукты желтого цвета:
R 2 NH + NaNO 2 + HCl → R 2 N-N=O + NaCl + H 2 O (12)
Нитрозамины являются сильными канцерогенами. Показана возможность синтеза нитрозаминов в желудке человека из содержащихся в пище и лекарственных препаратах вторичных аминов и нитритов Канцерогенное действие нитрозаминов основано на их способности алкилировать нуклеофильные центры ДНК, что приводит к онкогенным мутациям.
Первичные алифатические амины реагируют с азотистой кислотой с выделением газообразного азота. Реакция идет через образование неустойчивого первичного нитрозамина, который изомеризуется в диазогидроксид, превращающийся далее в соль диазония:
(13)
Дальнейший ход реакции зависит от природы углеводородного радикала.
Если R алифатический радикал, то соль диазония очень неустойчива и немедленно разлагается с образованием молекулы азота и карбокатиона, который затем взаимодействует с находящимися в реакционной среде нуклеофилами (например, с растворителем) или отщепляет протон и дает продукт элиминирования. Например, превращения катиона н-пропилдиазония могут быть представлены следующей схемой (Рис. 36):
Рис. 36. Превращение катиона н-пропилдиазония
Реакция не имеет препаративного значения. Процесс используется в аналитических целях для количественного определения первичных алифатических аминов, в том числе природных α-аминокислот, по объему выделяющегося азота.
Соли арилдиазония более устойчивы и могут быть выделены из реакционной смеси. Они являются высокореакционноспособными соединениями и широко используются в органическом синтезе.
Процесс получения ароматических диазосоединений называется диазотированием и выражается следующим суммарным уравнением.
ArNH 2 + NaNO 2 + 2HCl → ArN 2 + Cl - + NaCl + 2H 2 O (14)
Реакции солей арилдиазония можно разделить на два типа: реакции с выделением азота и реакции без выделения азота.
Реакции, протекающие с выделением азота. Этот тип реакций представляет собой замещение в ароматическом кольце, уходящей группой в котором является молекула азота N 2 (Рис. 37):
Рис. 37. Реакции солей арилдиазония
Реакции используются для введения различных заместителей в ароматическое кольцо.
Реакции, протекающие без выделения азота. Наиболее важной реакцией этого типа является азосочетание. Катион диазония обладает слабыми электрофильными свойствами и вступает в реакции электрофильного замещения с аренами, содержащими сильные электронодонорные заместители. При этом образуются азосоединения (Рис. 38):
Рис. 38. Реакции азосочетания
Азосоединения содержат длинную систему сопряженных связей и поэтому окрашены. Они используются как красители. Образование окрашенных соединений при взаимодействии солей арилдиазония с ароматическими аминокислотами (тирозин, гистидин) используется для их качественного и количественного определения.
Аминогруппа является сильным активирующим заместителем и ориентантом II рода.
Анилин легко бромируется бромной водой с образованием триброманилина (Рис. 39):
Рис. 39. Реакция бромирования анилина
В большинстве реакций электрофильного реакционноспособная аминогруппа предварительно защищается путем ацилирования. После проведения реакции ацильную защиту снимают кислотным или щелочным гидролизом (Рис. 40) :
Рис. 40. Реакция ацилирования
2.3. Методы получения аминов
2.3.1. Алкилирование аммиака и аминов
Аммиак взаимодействуют с алкилгалогенидами RX с образованием на первой стадии соли алкиламмония, которая с избытком аммиака дает алкиламин. Алкиламин, будучи более сильным нуклеофилом, чем аммиак, далее вступает в реакцию алкилгалогенидом с образованием продукта диалкилирования. Таким образом образуется смесь моно-, ди-, триалкиламинов и четвертичной аммониевой соли (Рис. 41):
Рис. 41. Реакции с алкилгалогенидом
Спирты алкилируют аммиак и амины в присутствии катализаторов дегидратации (Al 2 O 3 , SiO 2 ) при 300-500 0 C. В этом случае также образуется смесь продуктов алкилирования (Рис. 42) :
Рис. 42. Алкилирование аммиака и аминов спиртами
2.3.2. Восстановление азотсодержащих органических соединений
Нитросоединения могут быть восстановлены до первичных аминов. Реакция используется в основном для получения первичных ароматических аминов из доступных нитроаренов:
ArNO 2 ArNH 2 (15)
В качестве восстановителей используют водород в присутствии катализаторов (Ni, Pt, Pd), металл (Fe, Zn, Sn) и кислоту, соли металлов в низших степенях окисления (SnCl 2 , TiCl 3 ).
Нитрилы при восстановлении также дают первичные амины (Рис. 43):
RCN RCH 2 NH 2
[ H ]: H 2 / Ni ; LiAlH 4
Рис. 43. Восстановление нитрилов
Амиды карбоновых кислот восстанавливаются до аминов комплексными гидридами металлов. Из соответствующих амидов могут быть получены первичные, вторичные и третичные амины (Рис. 44):
Рис. 44. Восстановление амидов карбоновых кислот
Восстановительное аминирование альдегидов и кетонов (Рис. 45) :
Рис. 45. Восстановительное аминирование альдегидов и кетонов
2.3.3. Перегруппировка Гофмана
RCONH 2 + Br 2 + 2NaOH → RNH 2 + 2NaBr + CO 2 + H 2 O (16)
Используется для получения первичных аминов .
2.4. Биологически активные амины и их производные
Биологическую активность проявляют гетерофункциональные соединения, содержащие аминогруппу аминокарбоновые кислоты, аминоспирты, аминофенолы, аминосульфокислоты.
К ним можно отнести гормоны надпочечников (норадреналин, адреналин), щитовидной железы (тироксин, трийодтиронин), а также медиаторы ЦНС (ацетилхолин, ГАМК и др.), медиатор воспаления (гистамин) и другие соединения.
Некоторые аминокислоты и их производные могут подвергаться декарбоксилированию - отщеплению карбоксильной группы. В тканях млекопитающих декарбоксилированию может подвергаться целый ряд аминокислот или их производных: Три, Тир, Вал, Гис, Глу, Цис, Apr, Орнитин, SAM, ДОФА, 5-окситриптофан и др. Продуктами реакции являются СО 2 и амины, которые оказывают выраженное биологическое действие на организм (биогенные амины) (Рис. 46):
Рис. 46. Реакция получения биогенных аминов
Реакции декарбоксилирования необратимы и катализируются ферментами декарбоксилазами. Простетическая группа декарбоксилаз в клетках животных - пиридоксальфосфат. Некоторые декарбоксилазы микроорганизмов могут содержать вместо ПФ остаток пирувата - гистидиндекарбоксилаза Micrococcus и Lactobacilus, SAM-декарбоксилаза Е. coli и др. Механизм реакции напоминает реакцию трансаминирования с участием пиридоксальфосфата и также осуществляется путём формирования Шиффова основания и аминокислоты на первой стадии.
Амины, образовавшиеся при декарбоксилировании аминокислот, часто являются биологически активными веществами. Они выполняют функцию нейромедиаторов (серотонин, дофамин, ГАМК и др.), гормонов (норадреналин, адреналин), регуляторных факторов местного действия (гистамин, карнозин, спермин и др.).
Этаноламин (коламин) HOCH 2 CH 2 NH 2 является структурным компонентом сложных липидов. В организме образуется при декарбоксилировании аминокислоты серина .
Холин HOCH 2 CH 2 N + (CH 3 ) 2 гидроокись 2-оксиэтилтриметиламмония . Относят к витаминам группы В, хотя животные и микроорганизмы способны его синтезировать. Холин входит в состав фосфолипидов (например, лецитина, сфингомиелина), служит источником метильных групп в синтезе метионина. Из холина в организме животных синтезируется ацетилхолин один из важнейших химических передатчиков нервных импульсов. Холин является т.н. липотропным веществом предотвращает тяжёлые заболевания печени, возникающие при её жировом перерождении.
Холин - в медицине для лечения заболеваний печени применяют хлорид холина. Его вводят также в состав комбикормов сельскохозяйственным животных. Для аналитических целей используют способность холина давать плохо растворимые соли с фосфорновольфрамовой, платинохлористоводородной и некоторыми др. гетерополикислотами.
Ацетилхолин CH 3 COOCH 2 CH 2 N + (CH 3 ) 2 - посредник при передаче нервных импульсов (нейромедиатор). Накопление ацетилхолина в организме приводит к непрерывной передаче нервных импульсов и сокращению мускульной ткани. На этом основано действие нервнопаралитических ядов (зарин,табун), которые ингибируют действие фермента ацетилхолинэстеразы, катализирующего расщепление ацетилхолина.
Катехоламины дофамин, норадреналин, адреналин биогенные амины, продукты метаболизма аминокислоты фенилаланина (Рис. 47):
Рис. 47. Формулы катехоламинов
Дофамин обладает рядом физиологических свойств, характерных для адренергических веществ.
Дофамин вызывает повышение сопротивления периферических сосудов (менее сильное, чем под влиянием норадреналина). Он повышает систолическое артериальное давление в результате стимуляции α-адренорецепторов. Также дофамин увеличивает силу сердечных сокращений в результате стимуляции β-адренорецепторов. Увеличивается сердечный выброс. Частота сердечных сокращений увеличивается, но не так сильно, как под влиянием адреналина.
Потребность миокарда в кислороде под влиянием дофамина повышается, однако в результате увеличения коронарного кровотока обеспечивается повышенная доставка кислорода.
В результате специфического связывания с дофаминовыми рецепторами почек дофамин уменьшает сопротивление почечных сосудов, увеличивает в них кровоток и почечную фильтрацию. Наряду с этим повышается натрийурез. Происходит также расширение мезентериальных сосудов. Этим действием на почечные и мезентериальные сосуды дофамин отличается от других катехоламинов (норадреналина, адреналина и др.). Однако в больших концентрациях дофамин может вызывать сужение почечных сосудов.
Дофамин ингибирует также синтез альдостерона в коре надпочечников, понижает секрецию ренина почками, повышает секрецию простагландинов тканью почек.
Основной физиологический эффект мелатонина заключается в торможении секреции гонадотропинов. Кроме того, снижается, но в меньшей степени, секреция других тропных гормонов передней доли гипофиза кортикотропина, тиротропина, соматотропина.
Секреция мелатонина подчинена суточному ритму, определяющему, в свою очередь, ритмичность гонадотропных эффектов и половой функции. Синтез и секреция мелатонина зависят от освещённости избыток света тормозит его образование, а снижение освещённости повышает синтез и секрецию гормона .
Серотонин это вещество, являющееся химическим передатчиком импульсов между нервными клетками человеческого мозга и контролирующее аппетит, сон, настроение и эмоции человека.
Серотонин «руководит» очень многими функциями в организме. Например, очень интересны исследования его влияния на проявление боли. Доктором Виллисом доказано, что при снижении серотонина повышается чувствительность болевой системы организма, то есть даже самое слабое раздражение отзывается сильной болью.
Катехоламины физиологически активные вещества, выполняющие роль химических посредников в межклеточных взаимодействиях.
Адреналин оказывает стимулирующее воздействие на ЦНС. Он повышает уровень бодрствования, психическую энергию и активность, вызывает психическую мобилизацию, реакцию ориентировки и ощущение тревоги, беспокойства или напряжения, гегенируется при пограничных ситуациях.
Структурно близки к катехоламинам некоторые природные и синтетические биологически активные вещества, также содержащие аминогруппу в b-положении к ароматическому кольцу (Рис. 48):
Рис. 48. Формулы фенамина и эфедрина
Фенамин является стимулятором центральной нервной системы, снимает чувство усталости. Эфедрин алкалоид, обладающий сосудорасширяющим действием.
Производные п-аминофенола парацетамол и фенацетин лекарственные препараты, обладающие обезболивающим и жаропонижающим действием (Рис. 49):
Рис. 49. Формулы парацетамола и фенацетина
В настоящее время фенацетин рассматривается как вещество, возможно являющееся канцерогеном для человека.
п-Аминобензойная кислота и ее производные (Рис. 50):
Рис. 50. п-Аминобензойная кислота и ее производные
п-Аминобензойная кислота витаминоподобное вещество, фактор роста микроорганизмов; участвует в синтезе фолиевой кислоты (витамина В С ). Сложные эфиры п-аминобензойной кислоты вызывают местную анестезию.
Анестезин и новокаин применяются в виде растворимых в воде гидрохлоридов.
Сульфаниловая кислота (п-аминобензолсульфокислота) и сульфаниламиды. Амид сульфаниловой кислоты (стрептоцид) и его N-замещенные производные эффективные антибактериальные средства. Синтезировано более 5000 производных сульфаниламида. Наибольшую активность проявляют сульфониламиды, содержащие гетероциклические основания (Рис. 51):
Рис. 51. Формулы производных сульфаниловой кислоты
Антибактериальное действие сульфамидных препаратов основано на том, что они имеют структурное сходство с п-аминобензойной кислотой и являются ее атиметаболитами. Присутствующие в бактериальной среде сульфаниламиды включаются в процесс биосинтеза фолиевой кислоты, конкурируя с п-аминобензойной кислотой, и на определенной стадии блокируют его, что ведет к гибели бактерий. Сульфаниламиды не влияют на организм человека, в котором фолиевая кислота не синтезируется .
2.5. Акридон: получение, свойства и применение
Незамещенный акридон очень устойчивое в обычных растворителях желтое вещество; оно плохо растворимо и плавится при высокой температуре 354 ° C . Кристаллизуется в виде игл (Рис. 52):
Рис. 52. Формула акридона
Акридоны удобнее всего рассматривать как циклические винилоги амидов кислот. Они представляют собой ассоциированные, высоко плавящиеся и довольно слабо растворимые соединения, которые лучше всего перекристаллизовывать из пиридина или высококипящих растворителей. Химически они очень устойчивы, и при восстановлении их в акридины приходится преодолевать высокий энергетический барьер, следствием чего является последующая стадия восстановления с образованием акриданов. Процесс восстановления до акридана, однако, не имеет существенного значения, так как акриданы легко окисляются в акридины и более глубоко эта реакция уже не протекает. Существуют и другие пути превращения акридонов в акридины, не включающие непосредственного восстановления. Акридоны окрашены в кремовый или желтый цвет, многие сильно флуоресцируют. Хемилюминесценции и раздражающего действия у соединений акридона не наблюдается. Почти все акридоны (даже аминопроизводные) - чрезвычайно слабые основания. Акридон отличается от изомерных ему оксиакридинов отсутствием явно выраженных кислых и основных свойств. Спектр акридона тоже значительно отличается от спектров оксиакридинов. Молекулярный вес акридона был определен криоскопически в феноле; оказалось, что в этих условиях акридон мономерен; однако показано, что вещества такого типа могут состоять из коротких цепей молекул, соединенных водородной связью.
Акридон получается при окислении акридина дихроматом натрия в уксусной кислоте и при циклизации дифениламин-2-карбоновой кислоты (Рис. 53):
Рис. 53. Получение акридона
Натрием в этаноле он восстанавливается в акридан, который может быть окислен в акридин (Рис. 54):
Рис. 54. Получение акридана
Производные акридона находят применение в медицине, например циклоферон, обладающий антивирусными и иммуномоделирующими свойствами и способный подавлять развитие ряда микробов .
2.6. 9-аминоакридин: получение, свойства и применение
9-аминоакридин представляет собой светло-желтый порошок (Рис. 55):
Рис. 55. Формула 9-аминоакридина
Методы получения аминоакридинов более многочисленны, чем методы синтеза самого акридина. 9-аминоакридин можно получить несколькими способами. Один из них это получение его из 9-хлоракридина нагреванием с карбонатом аммония в присутствии фенола (Рис. 56):
Рис. 56. Получение 9-аминоакридин
9-аминоакридин может быть также получен из акридина по реакции с амидом натрия (реакция Чичибабина) (Рис. 57):
Рис. 57. Получение 9-аминоакридин по реакции Чичибабина
Кроме того, 9-аминоакридин может быть получен гидролизом 9-цианакридина (из акридина или 9-хлоракридина) до соответствующего амида кислоты, и последующим превращением амида в желаемый амин, а также разложением азида кислоты. При восстановлении хлоропроизводного водородом на Ni -Ренея образуется 9,10-дигидроакридин, окисление которого хромовой кислотой приводит к незамещенному акридину.
Синтез новых веществ в ряду аминоакридинов представляет собой большой практический интерес .
3. Заключение
Таким образом, амины это важный класс органических соединений, которые имеют свои характерные особенности.
Прежде всего, как стало ясно, реакции ацилирования, характерные для аминов, обладают многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). В результате чего можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и др. полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях.
Реакции алкилирования, которые также характерны для аминов, представляют собой последовательное замещение алкильными группами атомов водорода, находящихся у азота в первичных аминах, что приводит к образованию вторичных и третичных аминов. Введение алкильных групп легко достигается действием на амин соответственного галоидного алкила или алкилсульфата. Другой способ, по которому удается получать вторичные амины с значительно лучшими выходами, основан на способности металлических производных многих замещенных амидов типа RCONHR реагировать с галоидными алкилами. Из продукта алкилирования при гидролизе получается вторичный амин.
Еще один способ получения гомологов метиланилина заключается в нагревании галоидного алкила с большим избытком ароматического амина.
Новый способ получения моно- и диметильных производных заключается в применении метилового эфира р-толуолсульфоновой кислоты на соответствующий амин, а для получения вторичных аминов предложено взаимодействие азометинов с йодистыми алкилами, причем образуются соединения, которые по прибавлении воды или спирта расщепляются на вторичный амин и альдегид.
4. Список литературы
- Андрюшкова О.В., Козлова А.В.Органическая химия. Избранные разделы: учебное пособие. Новосибирск: Изд-во НГТУ, 2010.
- Артеменко А.И. Органическая химия. М., 2012.
- Артеменко А.И. Удивительный мир органической химии. М.: Высшая школа, 2011.
- Березин Б.Д., Березин Д.Б. Курс современной органической химии. Учебное пособие для ВУЗов. М.: Высшая школа, 2010.
- Иванов В.Г., Горленко В.А., Гева О.Н. Органическая химия М.: Мастерство, 2012.
- Ким К.Н. Органическая химия. Новосибирск, 2012.
- Левитина Т.П. Справочник по органической химии: Учебное пособие. - СПб: «Паритет», 2012.
- Нейланд О.Я. Органическая химия: Учебник для ВУЗов. М.: Высшая школа, 2012.
- Реутов О.И., Курц А.Л., Бутин К.П. Органическая химия: в 2 т. М.: Изд.МГУ, 2010.
- Скворцов А.В. Курс лекций по органической химии. Новосибирск: НГТУ, 2012.
- Шабаров Ю.С. Органическая химия: Учебник для ВУЗов.- 2-е изд.- М.: Химия, 2012.
EMBED PBrush

EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
EMBED ChemWindow.Document
Ароматические амины способны замещать водород аминогруппы на алкилы. Эта реакция приводит к вторичным и третичным аминам:
C 6 H 5 NH 2 + CH 3 I → C 6 H 5 -NH-CH 3 + CH 3 I → C 6 H 5 -N(CH 3) 2
Алкилированиеведут спиртами или хлоралаканами, в качестве катализаторов используют соли одновалентной меди в виде аммиачных комплексов. Важно, что процесс алкилирования является последовательно-параллельным. Это обусловлено тем, что образовавшийся амин, в свою очередь, способен реагировать с алкилирующим агентом. Состав продуктов зависит от соотношения реагентов.
2. Ацилирование ароматических аминов
При действии ацилирующих агентов (кислоты, ангидриды, хлорангидриды) водородные атомы аминогруппы замещаются на ацильные остатки.
Ацильные производные не обладают основными свойствами. Они обладают устойчивостью к окислителям и потому используются в качестве промежуточных веществ в реакциях аминов в присутствии окислителей (например, нитрование).
3. Синтез азометинов (оснований Шиффа)
При слабом нагревании ароматических первичных аминов с ароматическими альдегидами легко образуются так называемые основания Шиффа или азометины:

Под действием разбавленных кислот основания Шиффа гидролизуются до альдегида и амина.
4. Реакции аминов с азотистой кислотой
Первичные ароматические амины с азотистой кислотой при 0 – 5°С образуют соли диазония:

Вторичные амины при взаимодействии с азотистой кислотой образуют N-нитрозо-N-метиланилины:

Третичные амины с азотистой кислотой вступают в реакцию электрофильного замещения:

Важнейшие представители ароматических аминов
Анилин впервые был получен в результате перегонки индиго с известью (1826 г.). В 1842 г. его получил Зинин восстановлением нитробензола. В незначительных количествах содержится в каменноугольной смоле. В промышленности получают из нитробензола каталитическим гидрированием с медным катализатором в газовой фазе. Анилин в больших количествах идет на получение красителей, циклогексиламина, капролактама, пестицидов и др.
п-Толуидин широко применяется в производстве красителей, особенно фуксина.
N , N -диметиланилин применяется в производстве красителей и взрывчатых веществ.
Лекция 28. ДИАЗОСОЕДИНЕНИЯ. АЗОСОЕДИНЕНИЯ
Реакция диазотирования, условия проведения, механизм. Влияние заместителей в бензольном кольце на скорость реакции. Строение диазосоеиинений в зависимости от рН, таутомерные превращения. Химические свойства. Реакции, протекающие с выделением азота: нуклеофильное замещение диазониевой группы на гидроксил, алкоксигруппу, галогены. Механизм реакции. Реакции, протекающие без выделения азота. Условия реакции азосочетания с аминами и фенолами. Влияние заместителей на реакционную способность диазосоединения. Понятие об азокрасителях.
Реакция первичных ароматических аминов с азотистой кислотой приводит к образованию солей диазония (II. Гриси, 1858 г.). Эти соли имеют общую формулу [Аг-N≡N] + X - (где Х Cl, Br, NO 2 , HSO 4 , и т.д.):

Названия солей диазония образуются добавлением окончания -диазоний к названию радикала исходного ароматического соединения с указанием названия аниона, например фенилдиазоний хлорид или хлористый фенилдиазоний.
Лекции № 41-42
АМИНЫ
Амины можно рассматривать как производные аммиака, в котором атомы водорода замещены на углеводородные радикалы.
1. Классификация , изомерия, номенклатура
В зависимости от числа углеводородных радикалов, связанных с атомом азота,
различают первичные, вторичные и третичные амины, а также четвертичные
аммониевые соли.
|
RNH 2 |
RR / NH |
RR / R // N |
RR / R // R /// N + X - |
|
первичные амины |
вторичные амины |
третичные амины |
четвертичные аммониевые соли |
По типу гибридизации атома углерода, связанного с азотом выделяют следующие группы аминов.
К этой группе относятся алкиламины, а также алкенил- и алкиниламины, в которых кратная связь удалена от атома азота. Их объединяют под названием алифатические амины . В состав этой группы входят также циклические амины, содержащие атом азота в цикле, которые являются гетероциклическими соединениями.

К этой группе принадлежат производные алкенов с атомом азота у атома углерода, образующего двойную связь – енамины (виниламины) и амины, содержащие атом азота, связанный с ароматическим кольцом - ароматические амины (ариламины).
Названия аминов образуют, добавляя к слову амин названия связанных с атомом азота углеводородных радикалов.
В другом варианте номенклатуры за основу названия принимают название родоначальной структуры (самой длинной углеродной цепи, непосредственно связанной с атомом азота) с добавлением суффикса “амин”.

В этом случае вторичные и третичные амины называют как N-замещенные производные первичных аминов.

Если молекула содержит другие функциональные группы, обозначаемые в суффиксе, то аминогруппу обозначают префиксом “амино”.
![]()
Названия диаминов образуют от названий соответствующих двухвалентных радикалов или названия родоначальной структуры с добавлением суффикса “диамин”.

Многие ароматические амины имеют тривиальные названия.

Циклические амины называют, используя номенклатуру гетероциклических соединений или, добавляя к названию двухвалентного углеводородного радикала суффикс “имин”.

Для аминов характерна изометрия углеродного скелета, изомерия положения
аминогруппы и изомерия между первичными, вторичными и третичными аминами.
2. Алифатические амины
2.1. Методы получения.
1) Алкилирование аммиака и аминов.
Аммиак взаимодействуют с алкилгалогенидами RX и другими алкилирующими реагентами (алкилсульфатами, диалкилсульфатами) с образованием на первой стадии соли алкиламмония, которая в равновесной реакции с избытком аммиака дает алкиламин. Алкиламин далее вступает в реакцию c алкилгалогенидом с образованием продукта диалкилирования и т.д. Таким образом последовательно образуются триалкиламин и соль тетраалкиламмония.

Реакция используется в основном для синтеза третичных аминов и тетраалкиламмониевых солей, так как первичные и вторичные амины, будучи более сильными нуклеофилами, чем аммиак, реагируют далее, сами предпочтительно атакуя субстрат. Приемлемые выходы первичных аминов получают при использовании большого избытка аммиака, а вторичных аминов – при большом избытке первичного амина.
Спирты алкилируют аммиак и амины в присутствии катализаторов дегидратации (Al 2 O 3 , SiO 2) при 300-500 0 C. В этом случае также образуется смесь продуктов моно-, ди- и триалкилирования.

Метод используется для получения низших алифатических аминов в промышленности.
2) Синтез первичных аминов по Габриэлю

Алкилирование фталимида калия алкилгалогенидами с последующим щелочным гидролизом или гидразинолизом N-алкилфталимида позволяет получать первичные амины без примеси вторичных и третичных. Лучше использовать протекающий в мягких условиях гидразинолиз, приводящий к образованию не растворимого в реакционной среде циклического гидразида.
3) Восстановление азотсодержащих органических соединений.
Нитрилы при восстановлении дают первичные амины. В промышленности процесс осуществляют путем каталитического гидрирования.

В препаративных целях используют восстановление алюмогидридом лития.
![]()
Введение цианогруппы (например, путем нуклеофильного замещения) и ее восстановление – синтетический прием, позволяющий нарастить углеродную цепь на один атом С.
Амиды карбоновых кислот восстанавливаются до аминов алюмогидридом лития. Из соответствующих амидов могут быть получены первичные, вторичные и третичные амины.

Восстановление азотсодержащих производных альдегидов и кетонов – оксимов и гидразонов – дает возможность превращения карбонильных соединений в первичные амины.

Для восстановления используют каталитическое гидрирование, комплексные гидриды металлов (LiAlH 4).
Нитросоединения могут быть восстановлены до первичных аминов.
![]()
В качестве восстановителей чаще всего используют металл (Fe, Zn, Sn) и кислоту; алюмогидрид лития. В алифатическом ряду метод не находит широкого применения из-за ограниченной доступности алифатических нитросоединений по сравнению с ароматическими.
Восстановление азидов дает первичные амины.

Исходные азиды легко могут быть получены из алкилгалогенидов или сульфонатов путем нуклеофильного замещения.
4) Восстановительное аминирование карбонильных соединений .
Взаимодействие альдегидов и кетонов с аммиаком в присутствии восстановителя приводит к первичным аминам.

При использовании вместо аммиака первичного амина продуктом реакции будет вторичный амин.

Процесс протекает через промежуточное образование имина с его последующим восстановлением в амин.

Восстановительное аминирование с использованием в качестве восстановителя муравьиной кислоты называют реакцией Лейкарта-Валлаха. В качестве реагентов можно использовать формиат аммония или соответствующие соли аминов.
5) Синтез аминов путем перегруппировок.
Перегруппировка Гофмана:
RCONH 2 + Br 2 + 2NaOH ® RNH 2 + 2NaBr + CO 2 + H 2 O
Перегруппировка Курциуса:

Реакции подробно рассмотрены ранее (см. лек. №36) В результате образуются
первичные амины без примеси вторичных и третичных. При этом происходит
укорочение углеродной цепи на один атом С.
2.2. Физические свойства и строение
Алифатические амины – бесцветные вещества с неприятным запахом. Низшие амины – жидкости, хорошо растворимые в воде. По растворимости они превосходят спирты с близкой молекулярной массой. Это объясняется образованием между амином и водой водородных связей типа , прочность которых сравнительно велика в силу высокой основности атома азота. Температуры кипения и плавления у третичных аминов ниже, чем у первичных и вторичных с примерно одинаковой молекулярной массой, что связано с ассоциацией последних за счет образования межмолекулярных водородных связей.

Однако эти межмолекулярные водородные связи слабее, чем у спиртов, по
причине меньшей полярности связи N-Н по сравнению со связью О-Н. Вследствие
этого амины имеют более низкие температуры кипения, чем спирты с близкой
молекулярной массой.
Амины имеют пирамидальное строение. Величины углов R-N-R близки к
тетраэдрическому – 106-108 0 . Считается, что атом азота находится в
состоянии sp 3 -гибридизации, а четвертым лигандом является
неподелённая пара электронов (“фантом”-лиганд).
Третичные амины с разными углеводородными радикалами должны быть хиральными,
так как их молекулы не имеют плоскости симметрии. Однако за счет быстрой
пирамидальной инверсии, которая представляет собой акт рацемизации, их
невозможно выделить или зафиксировать в оптически активной форме.

Четвертичные аммониевые соли в случае разных заместителей существуют в виде пары устойчивых энантиомеров.

Спектральные характеристики.
В электронных спектрах аминов наблюдается поглощение в дальней УФ-области при 195-215 нм, что соответствует возбуждению неподеленной пары электронов азота (переход n® s* ).
В ИК-спектрах первичных и вторичных аминов наблюдаются полосы поглощения, связанные с валентными колебаниями связей N-H. Первичные амины характеризуются двумя полосами поглощения при ~3400 и ~3500 см -1 , вторичные амины – одной полосой при ~3500 см -1 .
В спектрах ПМР химический сдвиг протонов связи N-H находится в области 1-5 м.д. и значительно меняется в зависимости от концентрации, температуры и растворителя.
2.3. Химические свойства
Химическое поведение аминов определяется в основном наличием свободной пары электронов у атома азота, которая обусловливает их основные и нуклеофильные свойства. Реакции с участием связей N-H и N-C под действием оснований и нуклеофильных реагентов для аминов менее характерны.
Основные свойства
Алифатические амины являются одними из самых сильных незаряженных оснований (~ 10 - 11). Их водные растворы имеют щелочную реакцию.
R 3 N + H 2 O = R 3 NH + + OH -
С неорганическими кислотами амины образуют соли, которые в большинстве случаев хорошо растворимы в воде.
R 3 N + HX = R 3 N + X -
Основность аминов зависит от их строения и природы растворителя. Сравнение
основности в водных растворах показывает, что алкиламины являются более
сильными основаниями, чем аммиак. Вторичные амины превосходят по основности
первичные. Такой ряд основности согласуется с электронодонорным влиянием
алкильных групп (+I-эффект), которое способствует делокализации положительного
заряда в сопряженной кислоте (ионе аммония) и тем самым стабилизирует её в
большей степени, чем свободный амин. Однако это не объясняет уменьшения
основности при переходе от вторичных аминов к третичным.
|
NH 3 |
C 2 H 5 NH 2 |
(C 2 H 5) 2 NH |
(C 2 H 5) 3 N |
|
|
9,25 |
10,80 |
11,09 |
10,85 |
Вероятно, такое снижение основности связано с сольватацией. Сольватация молекулами воды триалкиламмониевого катиона затруднена присутствием трех гидрофобных алкильных групп и снижением возможности образования водородных связей.

Это предположение подтверждается тем, что в газовой фазе и в малополярных
растворителях третичные амины превосходят по основности вторичные.
Нуклеофильные свойства
а) Алкилирование
Примеры реакций алкилирования обсуждались при рассмотрении методов получения аминов.
б) Ацилирование
2RNH 2 + R / COX ® R / CONHR + RNH 3 X
2R 2 NH + R / COX ® R / CONR 2 + R 2 NH 2 X
Ацилирование аминов функциональными производными карбоновых кислот дает возможность получать вторичные и третичные амиды из первичных и вторичных аминов соответственно. Реакция подробно обсуждена ранее (лекция №36).
в) Взаимодействие с сульфонилхлоридами
Сульфонилхлориды взаимодействуют с аминами, давая сульфонамиды. Реакция с бензолсульфонилхлоридом лежит в основе пробы Гинсберга , позволяющей различать и разделять первичные, вторичные и третичные амины.
Сульфонамиды, образующиеся из первичных аминов, являются NH-кислотами и со щелочами дают растворимые в воде соли.

Вторичные амины дают сульфонамиды, которые не содержат кислого водорода и не растворяются в щелочах.

Третичные амины не реагируют.
г) Нитрозирование
Нитрозирование аминов происходит при взаимодействии с азотистой кислотой в кислой среде. Неустойчивую азотистую кислоту генерируют действием сильной кислоты на нитриты. Реакция протекает по-разному в зависимости от типа амина.
Первичные алифатические амины реагируют с образованием неустойчивых алкилдиазониевых солей, которые разлагаются с выделением газообразного азота и сложной смеси продуктов дезаминирования.
Образование солей диазония – сложный многостадийный процесс, который подробно будет рассмотрен на примере ароматических аминов.
Разложение катиона алкилдиазония приводит к образованию карбокатиона, который стабилизируется путем алкилирования присутствующих в реакционной среде нуклеофилов или путем отщепления протона с образованием алкена. Этим процессам может предшествовать изомеризация карбокатиона в энергетически более стабильный ион. Так, разложение катиона н -пропилдиазония в водном растворе наряду н-пропиловым спиртом дает изопропиловый спирт, а также продукт элиминирования - пропен.

Со вторичными аминами азотистая кислота образует нерастворимые в реакционной среде нитрозамины.
R 2 NH + NaNO 2 + HCl ® R 2 N-N=O + NaCl + H 2 O
Третичные амины в сильнокислой среде при комнатной температуре с азотистой кислотой не реагируют.
Нитрозирование аминов препаративного значения не имеет. Аналитическое значение этих реакций заключается в возможности качественно различить первичные, вторичные и третичные амины.
д) Галогенирование
Первичные и вторичные амины реагируют с гипогалогенитами с образованием N-галогенаминов.

N-галогенамины – сильные окислители и галогенирующие реагенты.
Окисление
Амины дают разнообразные продукты окисления, состав которых зависит от природы окислителя и строения амина.
Перекись водорода и надкислоты окисляют третичные амины до N-оксидов.
R 3 N + HOOH ® R 3 N + -O - + H 2 O
В случае первичных и вторичных аминов первоначально образующиеся N-оксиды перегруппировываются в производные гидроксиламина.

Такое окисление протекает сложно, так как гидроксиламины сами легко окисляются. В случае первичных аминов конечными продуктами окисления являются нитросоединения, например:
Первичные амины, в которых аминогруппа соединена с третичным атомом углерода, окисляются в нитросоединения перманганатом калия в водном ацетоне.

(R=Alk; R / =H, Alk)
Кислотные свойства
Первичные и вторичные алифатические амины являются очень слабыми NH-кислотами (pK а ~33-35). Их кислотные свойства проявляются при действии щелочных металлов или таких сильных основания, как металлоорганические соединения.

Образующиеся алкил- и диалкиламиды металлов – очень сильные основания. Диалкиламиды, содержащие вторичные или третичные алкильные радикалы (например, диизопропиламид лития), представляют интерес для органического синтеза как ненуклеофильные основания. Будучи сильными основаниями, они обладают низкой нуклеофильностью по причине стерических затруднений, возникающих при атаке электрофильных центров за исключением протона. Их используют в органическом синтезе для отрыва протона и генерирования карбанионов.
Расщепление гидроксидов тетраалкиламмония по Гофману
Гидроксиды тетраалкиламмония получают действием на галогениды оксида серебра.
2R 4 N + Br - + Ag 2 O + H 2 O ® 2R 4 N + OH - + 2AgBr
В растворах гидроксиды тетраалкиламмония полностью ионизированы и являются столь же сильными основаниями, как гидроксиды натрия и калия. При нагревании они претерпевают элиминирование с образованием алкена триалкиламина и воды.
RCH 2 CH 2 (CH 3) 3 N + OH - ® RCH=CH 2 + (CH 3) 3 N + H 2 O
При наличии в молекуле нескольких b -водородных атомов процесс протекает в направлении образования наименее замещенного алкена (по правилу Гофмана ).
Причина такой ориентации при отщеплении состоит в карбанионном характере переходного состояния, что способствует отщеплению наиболее кислого протона.

При протекании процесса по механизму Е2 с “Е1 СВ -подобным” переходным состоянием на атоме углерода возникает частичный отрицательный заряд. Переходное состояние (I), предшествующее образованию продукта по правилу Зайцева, оказывается дестабилизированным за счет +I-эффекта алкильных групп. В результате процесс преимущественно направляется через наименее дестабилизированное переходное состояние (II), ведущее к продукту элиминирования по Гофману.
3. Енамины
Енамины (виниламины) устойчивы в том случае, если при атоме азота нет атомов водорода. Такие енамины можно рассматривать как азотистые аналоги виниловых эфиров. В противном случае енамины нестабильны и перегруппировываются в имины, подобно тому, как енолы изомеризуются в карбонильные соединения.
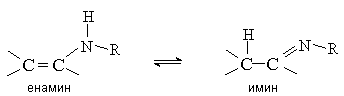
3.1. Методы получения
Основной метод получения енаминов – взаимодействие карбонильных соединений со вторичными аминами в присутствии кислотных катализаторов и средств, связывающих воду.
Реакция протекает по общему для присоединения азотистых оснований к карбонильной группе механизму.

Отщепление воды от интермедиата (III) приводит к образованию иммониевого иона (IV), который при отсутствии водорода у атома азота стабилизируется путем отщепления протона от b -углеродного атома.
3.2. Строение
Молекула енамина представляет собой р-p -сопряженную систему, строение которой можно отразить набором двух резонансных структур.
Таким образом, молекула енамина содержит два нуклеофильных центра – атом азота и атом углерода в b -положении, который несет частичный отрицательный заряд.
3.3. Химические свойства
а) Протонирование и гидролиз
Енамины являются слабыми основаниями. Их протонирование может протекать как по атому азота, так и по атому углерода. Образующаяся при протонировании по b -углеродному атому соль иммония гидролизуется, давая исходное карбонильное соединение и вторичный амин.
Гидролиз енаминов – процесс, обратный их образованию, и протекает по такому же механизму.
б) Алкилирование
Алкилирование енаминов алкилгалогенидами и другими алкилирующими реагентами протекает, как правило, по b -углеродному атому. Последующий гидролиз иммониевой соли приводит к карбонильному соединению.
Последовательность превращений – получение енамина из карбонильного соединения, алкилирование, гидролиз приводит к алкилированию исходного карбонильного соединения по a -положению и носит название реакция Сторка . Этот метод имеет преимущества перед алкилированием кетонов, так как требует более мягких условий и дает в основном продукты моноалкилирования.
Для проведения этой реакции чаще всего используют циклические амины – пирролидин, пиперидин, морфолин. Лучшие результаты достигаются при использовании активных галогенидов – аллил- и бензилгалогенидов, a -галогензамещенных производных простых и сложных эфиров. Например:

в) Ацилирование
При действии галогенангидридов и ангидридов кислот енамины дают продукты С-ацилирования. Последующий гидролиз приводит к дикарбонильному соединению.
Таким образом, последовательность реакция – получение енамина из карбонильного соединения, ацилирование, гидролиз – метод получения b -дикарбонильных соединений. Например:

4. Ароматические амины
4.1. Методы получения
1) Восстановление ароматических нитросоединений
![]()
Для восстановления в препаративных целях используют металл (Fe, Zn, Sn) и кислоту, соли металлов в низших степенях окисления (SnCl 2 , TiCl 3), сульфиды щелочных металлов, в промышленности применяют в основном каталитическое гидрирование. См. также лекцию №40.
2) Алкилирование

Реакция аналогична алкилированию алифатических аминов. В качестве алкилирующих реагентов используют алкилгалогениды, алкилсульфаты, спирты.
3) Арилирование
Галогенарены реагируют с аммиаком и аминами в жестких условиях. Процесс катализируется медью и ее соединениями.
Реакция замещения галогена протекает легче при наличии в орто - и пара -положениях электроноакцепторных групп (NO 2 , CN).
Галогенарены взаимодействуют с ариламинами в присутствии меди с образованием диариламинов (реакция Ульмана).

4.2. Физические свойства и строение
Ароматические амины – бесцветные жидкости или твердые вещества. При хранении быстро темнеют вследствие окисления кислородом воздуха.
Аминогруппа и ароматическое кольцо образуют сопряженную систему. Аминогруппа проявляет электронодонорные свойства за счет +М-эффекта.

Ароматические амины обладают сильными электронодонорными свойствами, на что указывают низкие энергии ионизации (для анилина 7,7 эВ, для фенола 8,4 эВ).
4.3. Химические свойства
Для ариламинов характерны реакции с электрофильными реагентами. Местом атаки электрофила может быть атом азота или ароматическое кольцо.
Основные свойства
Ароматические амины обладают меньшей основностью, чем алифатические амины и аммиак (~ 3 – 5). Причина низкой основности ариламинов – стабилизация свободного амина за счет делокализации неподеленной пары электронов азота по ароматическому кольцу и потеря энергии стабилизации при нарушении сопряженной системы в результате протонирования.
Дифениламин и трифениламин имеют еще большие возможности для делокализации пары электронов азота, что приводит значительному снижению основности. Трифениламин практически не обладает основными свойствами.
п-CH 3 C 6 H 4 NH 2п-O 2 NC 6 H 4 NH 2
C 6 H 5 NH 2
(C 6 H 5) 2 NH
0,78
м-O 2 NC 6 H 4 NH 2
Реакции с С-электрофилами
Важнейшими реакциями этого типа являются алкилирование и ацилирование, которые протекают по атому азота и аналогичны реакциям алифатических аминов. Ароматические амины менее реакционноспособны из-за меньшей основности атома азота.
Реакции ароматического электрофильного замещения
а) Галогенирование
Аминогруппа является сильным активирующим заместителем и ориентантом I рода. Галогенирование свободных аминов протекает очень легко и часто приводит к полигалоидпроизводным. Например, анилин при действии бромной воды мгновенно превращаются в нерастворимое 2,4,6-трибромпроизводное.

Для получения моногалогенпроизводных активирующее действие аминогруппы снижают путем ацилирования. После снятия ацильной защиты путем гидролиза получают свободный амин.


б) Нитрование
При нитровании нитрующей смесью амины окисляются. Кроме того, из-за солеобразования по аминогруппе образуется м-изомер (-NH 3 + - ориентант II рода).

Для введения нитрогруппы в орто - или пара -положение к аминогруппе последнюю защищают ацилированием. Варьируя условия реакций (температуру, нитрующие агенты), можно проводить нитрование региоселективно.

После снятия ацетильной защиты получают свободные орто - и пара -нитроанилины.
в) Сульфирование
Сульфированием ароматических аминов получают аминосульфокислоты. В 90-100%-ной серной кислоте или олеуме амин полностью находится в протонированной форме. Аммониевая группа NH 3 + как сильный электроакцепторный заместитель вызывает резкое замедление реакции сульфирования и ориентирует замещение в мета -положение.

Для получения орто- и пара -аминобензолсульфокислот используют “метод запекания”. Процесс осуществляют при длительном нагревании гидросульфатов ароматических аминов при 100-200 о С в сухом виде или в высококипящих растворителях. При температуре около 100 о С образуется практически чистый орто -изомер (ортаниловая кислота, продукт кинетического контроля), а при 180-200 о С - пара -изомер (сульфаниловая кислота, продукт термодинамического контроля).

Нитрозирование
Первичные ароматические амины с азотистой кислотой образуют относительно устойчивые соли арилдиазония.
ArNH 2 + NaNO 2 + 2HCl ® ArN 2 + Cl - + NaCl + 2H 2 O
Эту реакцию называют диазотированием (см.лек. №43).
Вторичные ароматические амины при нитрозировании дают N-нитрозамины.
ArNHR + NaNO 2 + HCl ® Ar-N(R)-N=O + NaCl + H 2 O
Третичные ариламины дают продукты нитрозирования в пара-положение ароматического кольца.
2. Ацилирование и алкилирование аминов
Третичные амины отличаются от первичных и вторичных аминов отсутствием способных к замещению атомов водорода, связанных с азотом. Это различие ясно проявляется при действии ацилирующих и алкилирующих средств; из первичных и вторичных аминов при ацилировании обычно получаются замещенные амиды, тогда как третичные амины выделяются в неизмененном состоянии после прибавления воды или водной щелочи. Атомы водорода в аминогруппе первичных и вторичных аминов могут быть замещены в определенных условиях алифатическим или ароматическим радикалом, или же остатками –СONH 2 , -С1, -Вг и -NO 2 . Эти реакции вкратце рассматриваются ниже.
Ацилирование
Способы, применяемые для ацилирования, могут быть в основном разделены на следующие группы: нагревание аминов с кислотами, взаимодействие аминов с хлорангидридами, бромангидридами или ангидридами кислот и реакция аминов со сложными эфирами, или даже с амидами кислот, дающая обычно худшие результаты.
Первый из этих способов состоит в нагревании амина с избытком соответствующей карбоновой кислоты.
Аналогичным путем получаются высшие гомологи ацетанилида. Этот способ часто применяется для идентификации одноосновных кислот. Интересно отметить, что муравьиная кислота значительно легче, чем ее гомологи, превращается в замещенные формамиды по этому способу. Форманилид легко образуется при нагревании 50%-ной водной муравьиной кислоты с анилином.
Для ацетилирования аминов рекомендуется также пользоваться тиоуксусной кислотой. Преимущество этого способа состоит в том, что ацетилирование анилина и его гомологов протекает в этом случае на холоду. Реакция протекает с выделением сероводорода.
Более удобный и распространенный способ получения ацилированных аминов заключается в применении хлорангидридов или ангидридов кислот. Хлорангидрид кислоты реагирует с избытком амина с образованием ацилированного производного и солянокислой соли амина.
Отделение солянокислой соли от ацилированного производного амина основано на их различной растворимости. Обычно реакцию ведут в таком растворителе, в котором соль амина нерастворима. Кроме того, если ацилированный амин нерастворим в воде, солянокислую соль легко удалить промыванием реакционной смеси водой
Если хлорангидрид кислоты сравнительно устойчив к действию воды и холодного водного раствора щелочи, введение ацильной группы может быть осуществлено по способу Шоттена и Баумана. Амин суспендируют в приблизительно 10%-ном водном растворе щелочи и обрабатывают хлорангидридом кислоты, взятым в 1,25-1,5-кратном против теории количестве. При этом реакционную смесь перемешивают или взбалтывают, пока большая часть хлорангидрида не прореагирует. Избыток хлорангидрида разлагают слабым нагреванием реакционной смеси. Образовавшееся трудно растворимое ацильное производное отфильтровывают, промывают водой до полного удаления щелочи и перекристаллизовывают из подходящего растворителя. Важно, чтобы в процессе реакции водный раствор-все время обладал щелочной реакцией. Этот метод с успехом применяется для хлорангидридов ароматических кислот, арилсульфоновых кислот и пирослизевой кислоты. Следует отметить, что сульфонильные производные первичных аминов растворимы в щелочах, а сульфонильные производные вторичных аминов нерастворимы. На этом основан способ распознавания и разделения первичных и вторичных аминов.
К другим способам ацилирования относятся действие хлорангидрида кислоты на эфирный раствор амина с суспендированным в нем углекислым калием, а также ацилирование в пиридине.
Для ацетилирования первичных ароматических аминов в лабораторных условиях целесообразно пользоваться уксусным ангидридом. Реакция между уксусным ангидридом и анилином или его гомологами протекает очень легко и обычно осуществляется прибавлением уксусного ангидрида к смеси амина примерно с 5-кратным по объему количеством воды. При реакции происходит выделение тепла и смесь быстро густеет, вследствие выделения ацетильного производного. Если амин обладает сравнительно высоким молекулярным весом, обычно удобнее перед прибавлением уксусного ангидрида смешивать основание с разбавленной уксусной кислотой. Применение спирта в качестве растворителя при ацетилировании аминов уксусным ангидридом на холоду имеет то преимущество, что избыток ангидрида легко можно удалить 1-2-кратным выпариванием со спиртом. Ацетилирование уксусным ангидридом в водной или спиртовой среде не дает удовлетворительных результатов при первичных ароматических аминах, содержащих в ядре отрицательные заместители.
Вышеупомянутый способ ацетилирования обладает тем преимуществом, что при этом не наблюдается образования диацетильных производных, что имеет место при применении неразбавленного уксусного ангидрида, например, в результате нагревания 10 г анилина с 40 г уксусного ангидрида в течение 1 часа продукт реакции представляет собой смесь, содержащую 10 г диацетиланилина и 5,6 г ацетанилида18. Наличие заместителей, например -СНз, -NO 2 , -С1, -Вг, в о-положении к аминогруппе благоприятствует образованию диацетильных производных. При нагревании о-толуидина с 4-кратным по весу количеством уксусного ангидрида с обратным холодильником получается диацетил-о-толуидин с прекрасным выходом.
Наличие в ядре ароматического амина нитрогруппы и, в меньшей степени, хлора или брома замедляет реакцию ацетилирования при комнатной температуре. Это явление становится особенно заметным при накоплении отрицательных групп в молекуле. При стоянии раствора 2, 4, б-триброманилина в избытке уксусного ангидрида при комнатной температуре в течение 2 недель ацетильное производное не образуется. Присутствие небольшого количества концентрированной серной кислоты очень сильно катализирует процесс ацетилирования, из 1 в 2, 4, 6-триброманилина в 20 г уксусного ангидрида в присутствии двух капель концентрированной серной кислоты при стоянии в течение 10 мин. при комнатной температуре получается чистый 2, 4, 6-трибромацетанилид. Для выделения продукта реакционную смесь выливают в воду.
Наилучший способ получения ацетильных производных низших алкнланилинов состоит в перегонке смеси равных объемов амина и уксусного ангидрида; выше 200° перегоняется ацетильное производное в довольно чистом состоянии. Многие из этих соединений кристаллизуются при охлаждении.
Третичные амины, благодаря своему строению, не способны к образованию амидов при взаимодействии с хлорангидридами или ангидридами кислот. Однако они могут давать с хлорангидридами продукты присоединения, которые обычно разлагаются при действии воды с образованием исходного амина. Например, продукт присоединения 1 моля пиридина к 1 молю хлористого ацетила при действии спирта превращается в солянокислый пиридин и уксусноэтиловый эфир. Хлористый диаллил также образует продукт присоединения к пиридину. Кроме того, описаны соединения, образующиеся при взаимодействии хлористого бензоила и хлористого ацетила с триэтиламином, пиридином, диметиланилином и некоторыми другими третичными аминами. Продукты присоединения триметиламина к арилсульфохлоридам сравнительно стойки к действию воды и дают хлороплатинаты и хлораураты.
Образование производных мочевины
Соли первичных и вторичных аминов с циановой кислотой более или менее легко изомеризуются с образованием замещенных производных мочевины
RNH 2 HCNO --> RNHCONH 2
Эта реакция аналогична превращению циановокислого аммония в мочевину. Следующие примеры иллюстрируют применение этого способа. Первичные и вторичные амины легко реагируют с эфирами изоциановой кислоты с образованием производных мочевины. Обычно для этой цели применяют фенилизоцианат, который нагревают с эквимолекулярным количеством амина в каком-либо не содержащем гидроксила растворителе, например в петролейном эфире.
Совершенно необходимо предохранять реакционную смесь от доступа влаги и применять лишь тщательно обезвоженные растворитель и амин, так как фенилизоцианат реагирует с водой с образованием дифенилмочевины. α-Нафтилизоцианат более удобен в этом отношении, чем фенилизоцианат, так как он менее чувствителен к действию воды и поэтому при реакции образуется меньшее количество нежелательных побочных продуктов.
Фенилизотиоцианат (фенилгорчичное масло) реагирует с аминами аналогичным образом с образованием соответственных производных тиомочевины. Реакция эта осуществляется в тех же условиях, как и с фенилизоцианатом.
Алкилирование первичных и вторичных аминов
Последовательное замещение алкильными группами атомов водорода, находящихся у азота в первичных аминах, ведет к образованию вторичных и третичных аминов. Введение алкильных групп легко достигается действием на амин соответственного галоидного алкила или алкилсульфата. Состав конечного продукта реакции зависит в значительной степени от относительных количеств взятых в реакцию компонентов, а также и от условий опыта, и обычно очень трудно получить при алкилировании только одно из возможных производных амина и поэтому продукт реакции, как правило, представляет собой смесь вторичного и третичного аминов наряду с значительным количеством непрореагировавшего первичного амина, а часто с примесью некоторого количества соли четвертичного аммониевого основания. Получение сложной смеси при применении галоидного алкила является результатом образования при реакции галоидоводорода, который дает соли с находящимися в реакционной смеси аминами. Распределение галоидоводорода между аминами зависит от их относительной основности, их сравнительного количества, а также от растворимости солей аминов в реакционной смеси. При алкилировании ароматических аминов выделяющийся осадок обычно содержит значительное количество соли исходного амина, а в растворе остается алкилированный амин, который вступает в дальнейшую реакцию с галоидным алкилом. Такие затруднения могут быть преодолены, по крайней мере, до известной степени проведением алкилирования в присутствии веществ, способных связывать образующийся галоидозодород, например, углекислой или двууглекислой соли щелочного металла. Для выделения вторичных аминов, наряду с вышеупомянутыми способами, обычно, за исключением некоторых особых случаев, пользуются способностью вторичных аминов образовывать нитрозамины. При восстановлении нитрозаминов оловом с соляной кислотой или при нагревании их с минеральными кислотами получаются чистые вторичные амины. Другой способ, по которому удается получать вторичные амины с значительно лучшими выходами, основан на способности металлических производных многих замещенных амидов типа RCONHR реагировать с галоидными алкилами. Из продукта алкилирования при гидролизе получается вторичный амин
Для этой цели удобно пользоваться ацетанилидом и его гомологами. Кроме того, применялись и формильные производные первичных ароматических аминов, а также арилсульфонильные производные первичных аминов.
Другой способ получения гомологов метиланилина заключается в нагревании галоидного алкила с большим избытком ароматического амина. По окончании реакции избыток ароматического амина осаждают прибавлением водного раствора хлористого цинка. Этот способ применялся для получения многих алкиланилинов с вполне удовлетворительными результатами. Таким же путем могут быть получены алкиланилины, содержащие третичную алкильную группу. Для алкилирования аминов также можно пользоваться ди-алкилсульфатами. Впрочем, обычно, этот способ ограничивается применением имеющегося в продаже диметилсульфата. Алкилирование по этому способу проводится в индиферентном растворителе или в присутствии водной щелочи, причем последняя модификация имеет более широкое применение. Вместо диалкилсульфатов можно1 применять эфиры арилсульфоновых кислот. Спирты вступают в реакцию с солями первичных ароматических аминов, примерно, при 200° с образованием моно- и диалкилариламинов. Эта реакция имеет применение в промышленности; для получения метиланилина нагревают при 180° смесь 55 частей солянокислого анилина и 16 частей метилового спирта. Для получения диметиланилина смесь 80 частей анилина, 78 частей метилового спирта и 8 частей серной кислоты нагревают в автоклаве до 235°. В лабораторных условиях можно вместо серной кислоты пользоваться другим катализатором, например йодом. Еще более активным катализатором в этой реакции является смесь порошкообразной меди с бромистым натрием или смесь галоидных солей меди и натрия Вторичные амины могут быть также получены восстановлением. Эта реакция может быть осуществлена электролитическим путем, действием цинковой пыли и водной щелочи натрия в спиртовой среде или муравьиной кислоте.
Новый способ получения метальных производных α- и β-нафтиламинов предложен Родионовым и Введенским. Для получения моно- и диметильных производных применяется действие метилового эфира р-толуолсульфоновой кислоты на соответствующий амин.
Другой интересный способ получения вторичных аминов основан на взаимодействии азометинов с йодистыми алкилами, причем образуются соединения, которые по прибавлении воды или спирта расщепляются на вторичный амин и альдегид.
Арилирование первичных и вторичных аминов
Введение в аминогруппу ароматического остатка обычно сопряжено с некоторыми затруднениями, вследствие малой реакционноспособности галоида в ароматических соединениях. Например, хлорбензол и бромбензол не вступают в реакцию с анилином в условиях, аналогичных применяемым для получения этиланилина. Впрочем, в присутствии медной бронзы или йодистой меди эта реакция протекает более гладко.
При взаимодействии третичных аминов с йодистыми алкилами образуются соли четвертичных аммониевых оснований. Общий способ получения таких соединений состоит в смешении обоих компонентов, иногда в каком-либо подходящем растворителе. Реакция протекает при комнатной температуре или при нагревании по схеме.
R""R"R"N + RHal -> R""R"R"RNHal
Реакция образования солей четвертичных аммониевых оснований часто применяется для идентификации третичных аминов, причем в качестве реактива наибольшее применение имеет йодистый метил. Рекомендуется также пользоваться для этой цели метиловым эфиром р-толуолсульфоновой кислоты. Ниже приводятся общие условия реакции для получения р-толуол-сульфоновокислых солей четвертичных аммониевых оснований.
При взаимодействии третичных аминов с йодистыми алкилами образуются соли четвертичных аммониевых оснований. Общий способ получения таких соединений состоит в смешении обоих компонентов, иногда в каком-либо подходящем растворителе. Реакция протекает при комнатной температуре или при нагревании по схеме
R""R"R"N + RHal -> R""R"R"RNHal
Вместо галоидных алкилов можно пользоваться диалкилсульфатами или алкиловыми эфирами ароматических сульфоновых кислот, причем получаются сернокислые или арилсульфоновокислые соли соответствующих четвертичных аммониевых оснований.
Соли четвертичных аммониевых оснований образуются не только в результате взаимодействия галоидных алкилов или эфиров ароматических сульфоновых кислот с третичными амидами, но и при действии эфиров йодуксусной кислоты на некоторые амины. Легче других вступают в эту реакцию бензилпиперидин, алифатические третичные амины и хинолин. В некоторых случаях для получения четвертичных аммониевых солей.
Легкость образования четвертичных аммониевых солеи сильно зависит от характера исходных соединений.
Наличие заместителей в о-положении к аминогруппе оказывает замедляющее действие на скорость реакции, что ясно видно из сравнения констант, для диметил-о-, -т- и р-толуидина, а также для хинолина и изохинолина. Это явление еще более резко выражено при наличии двух заместителей в о-положении к аминогруппе.
Например, третичные амины (III) и (IV) не реагируют с йодистым метилом при 100°, тогда как изомерные им амины, обладающие другим строением, сравнительно легко образуют четвертичные аммониевые соли

Диметилмезидин (V) и диметиламинопентаметилбензол (VI) также не способны к образованию четвертичных аммониевых соединений
N-метилакридиния), и осадок (5-окси-N-метилакридан) окисляют хромовым ангидридом. Акридон сульфируется и нитруется в положения 3 и 3,7, а при бромировании дает 2,3-дибромпроизводное. Мною апробирован метод получения акридона из фенилантраниловой кислоты. Выбранная реакция принадлежит к реакциям замыкания цикла. ОБЗОР ЛИТЕРАТУРЫ Реакции замыкания цикла. Типы реакций. Реакции замыкания...





Получаемый ПВХ отличается высокой полидисперсностыо и широким молекулярно-массовым распределением. Достоинства полимеризации в массе: высокая чистота полимера, его повышенные электроизоляционные свойства, прозрачность изделий. Производство поливинилхлорида в суспензии Большая часть ПВХ производится суспензионным методом, обеспечивающим высокое качество полимера (со сравнительно узким...

И, конечно же, за многими другими, которые будут получены, - будущее. В этом направлении и работают многие НИИ и исследователи. Аспекты поиска новых лекарств, изыскание новых лекарственных веществ состоит из трех основных этапов: химический синтез, установление фармакологической активности и безвредности (токсичности). Такая стратегия поиска с большой затратой времени, реактивов, животных, труда...





